【学习笔记】生物信息学基础知识+序列比对初步了解(一)
文章目录
- DNA和RNA的组成
- 基 因
- 蛋白质
- 中心法则
- DNA的复制
- DNA到mRNA转录
- 蛋白质的剪接
- 蛋白质的折叠
- 突变与多态性
- 组 学
- 转录组
- 蛋白质组
- 代谢组
- 组学数据简介
- 表观遗传
- 复杂生物网络
- 影像组
- 主要研究内容介绍
- 多序列比对
- 全局序列比对
- 动态规划算法(Dynamic Programming Algorithm)
- 分治法(Divide and Conquer Methods)
- SP方法(Sum of Pairs Methods)
- 累进方法(Progressive Methods)
- 迭代方法(Iterative Methods)
- 遗传算法(Genetic Algorithms)
- 局部序列比对
- 由概形分析(Profile Analysis)
- 区块分析(Block Analysis)
- 统计学方法(Statistical Methods)
- 穿插内容:mooc内容山东大学
- 1.序列定义及分类:蛋白质序列&核酸序列
- 2. 序列相似性
- 3. 一致度和相似度
- 4. 替换记分矩阵
- 总结:【BLAST矩阵】 为DNA序列最广泛采用的矩阵
- 总结:【PAM矩阵】为蛋白质序列比较中最广泛使用的积分方法之一
- 5.两个序列的长度相同计算一致度与相似度
- 6.两个序列的长度不同计算一致度与相似度
- 比较两个序列的方法:打点法&序列比对法
- (1)打点法:
- 两个序列打点判断是否相似
- 序列自己和自己打点寻找串联重复序列
- 打点法在线软件
- (2)序列比对法:
- 全局比对算法:Needleman-Wunsch算法
- 局部比对算法:Smith-Waterman算法
- 总结如何计算相似度
- `注意:不管两条序列长度是否相同,都要先对它们做全局比对。`
- `都是要除以全局比对的长度来得到一致度和相似度`
Bioinformatics is an study field that develops computational methods and software tool for understanding biological and medical data.
生物信息学(Bioinformatics)是研究生物信息的采集、处理、存储、传播,分析和解释等各方面的学科,也是随着生命科学和计算机科学的迅猛发展,生命科学和计算机科学相结合形成的一门新学科。它通过综合利用生物学,计算机科学和信息技术而揭示大量而复杂的生物数据所赋有的生物学奥秘。
DNA和RNA的组成
DNA和RNA是由具有相似化学结构的多聚体链组成的
DNA为脱氧核糖核酸;RNA为核糖核酸。
每个单元称为一个核甘酸(nucleotides)
核甘酸的组成
一个磷酸盐(phosphate)
一个环状的核糖(sugar)
一个环状的碱基
DNA由以下四种核甘酸编码而建立模式。 AGCT
腺嘌呤(Adenine)
鸟嘌呤(Guanine)
胞嘧啶(Cytosine)
胸腺嘧啶(Thymine)RNA的编码中尿嘧啶(Uracil)代替了胸腺嘧啶(Thymine)
DNA由以下四种核甘酸编码而建立模式。 AGCU
腺嘌呤(Adenine)
鸟嘌呤(Guanine)
胞嘧啶(Cytosine)
胸腺嘧啶(Thymine)
Chargaff规则
A(腺嘌呤)—T(胸腺嘧啶)或U(尿嘧啶)
C(鸟嘌呤)—G(胞嘧啶)
DNA的双螺旋结构
DNA分子的方向性:5’→3’
RNA链通常是单链,但可借助不同部位的互补和反序互补片断的耦合,形成一些二级结构
RNA类型:
Messenger RNA (mRNA):信使RNA是由DNA经剪接而成,携带遗传信息的能指导蛋白合成的一类单链核糖核酸。
Ribosomal RNA (rRNA):核糖体RNA(rRNA)是组成核糖体的主要成分,核糖体是合成蛋白质的工厂。
与蛋白质结合而形成核糖体,其功能是在mRNA的指导下将氨基酸合成为肽链
Transfer RNA (tRNA):具有携带并转运氨基酸功能的一类小分子核糖核酸。(tRNA上的是反密码子)
转录(Transcription)是遗传信息从DNA流向RNA的过程。 即以双链DNA中的确定的一条链(模板链用于转录,编码链不用于转录)为模板,以A,U,C,G四种核糖核苷酸为原料,在RNA聚合酶催化下合成RNA的过程。
基 因
DNA上具有特定功能的一个片断,负责一种特定性状的表达。
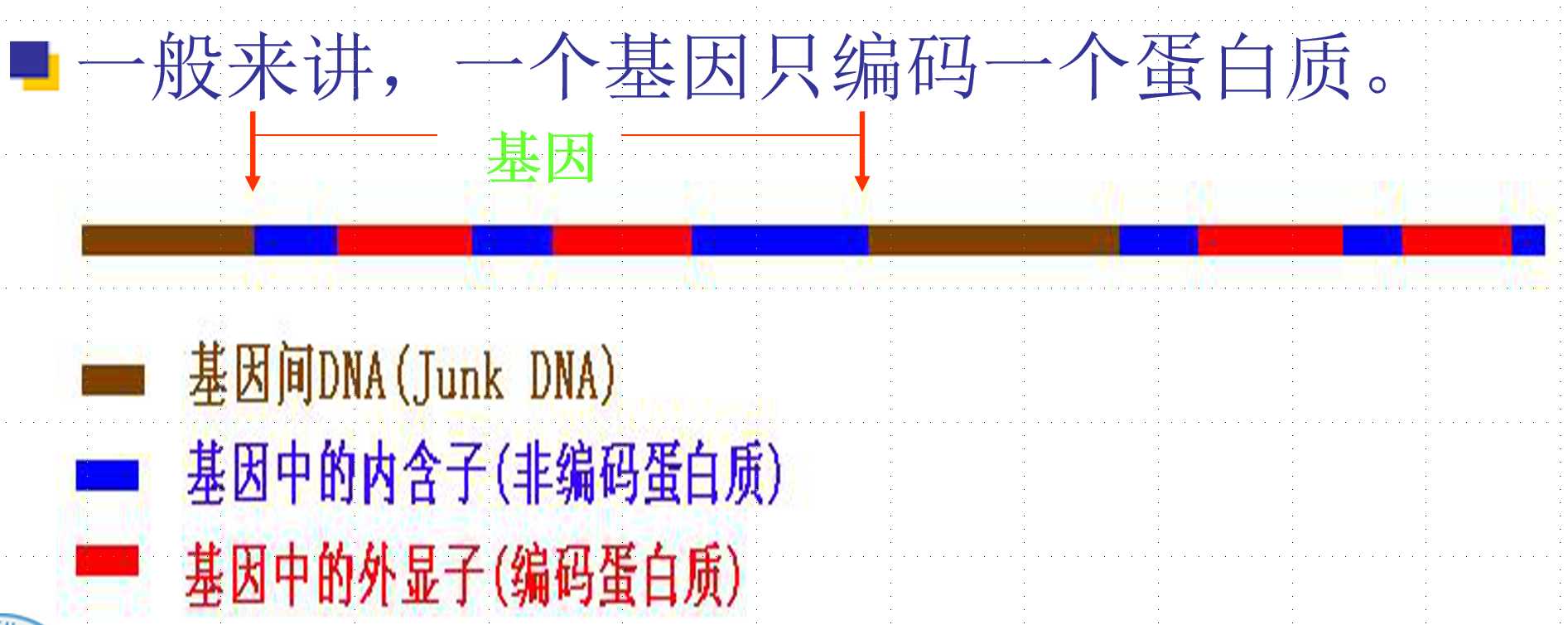
在DNA序列的编码区,每三个核甘酸翻译成蛋白质中一个特定的氨基酸。
每个核甘酸三元组称为一个密码子(codon)
核甘酸组成的三元组的排列共有4^3=64个密码子,其中有3个终止密码子UAA、UAG和UGA,其余61个密码子编码20种氨基酸,所以不同的密码子可能表示同一种氨基酸。
蛋白质
蛋白质是由20种氨基酸组成的线性多聚体,氨基酸之间由肽键相连接。
蛋白质是有方向的一维链,带氨基的一头称为N端(N’),另一头带羧基称为C端(C’)
构成蛋白质的氨基酸的数目从20到5000个不等;平均长度为350个氨基酸。


蛋白质结构![]() ](https://imgchr.com/i/BfY4fA)
](https://imgchr.com/i/BfY4fA)
中心法则
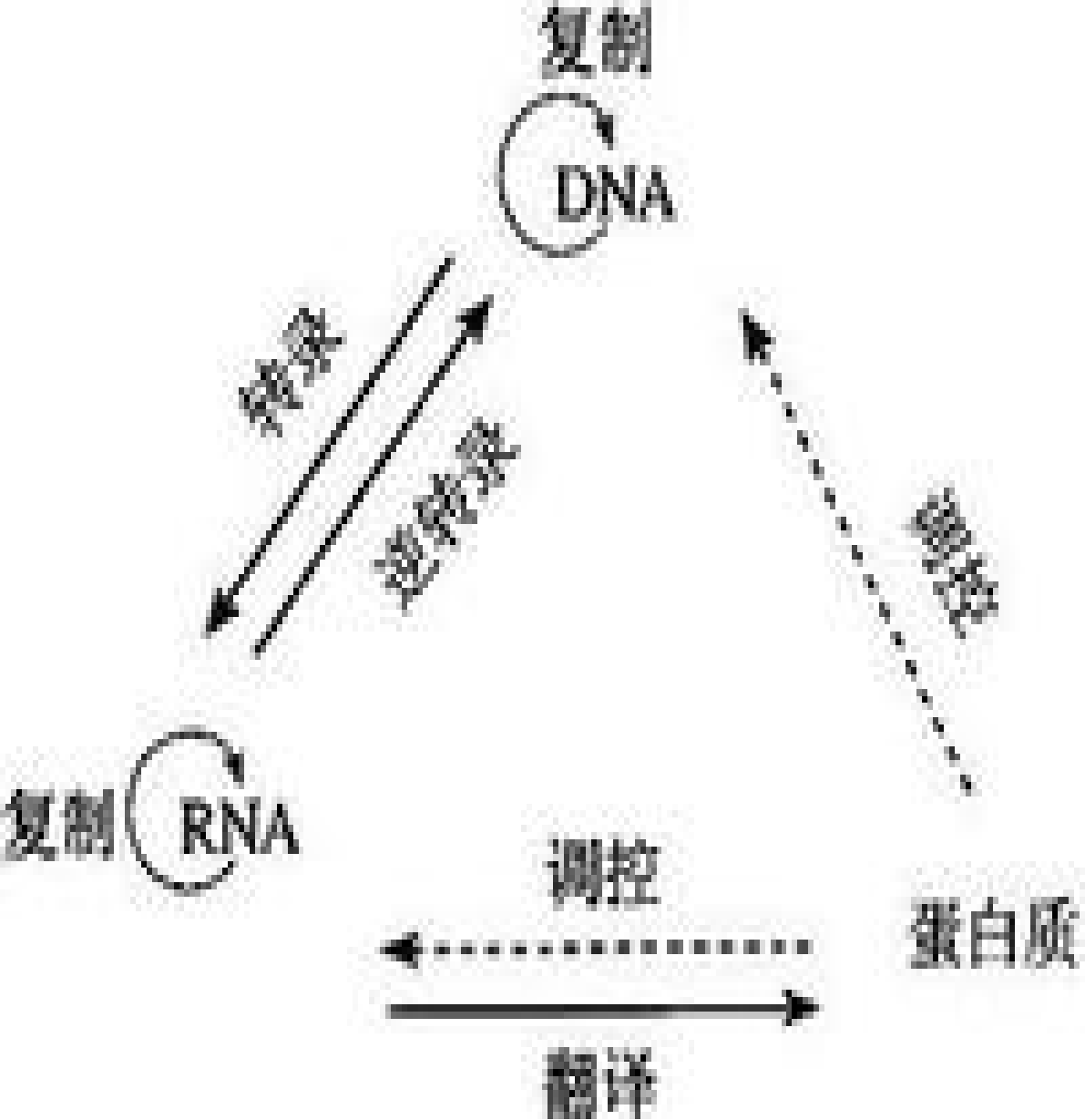
中心法则是1958年由克里克(Crick)提出的遗传信息传递的规律,包括由DNA到DNA的复制、由DNA到RNA的转录和由RNA到蛋白质的翻译等过程。(20世纪70年代逆转录酶的发现,表明还有由RNA逆转录形成DNA的机制,是对中心法则的补充和丰富。)
中心法则(genetic central dogma):是指遗传信息从DNA传递给RNA,再从RNA传递给蛋白质,即完成遗传信息的转录和翻译的过程。也可以从DNA传递给DNA,即完成DNA的复制过程。这是所有有细胞结构的生物所遵循的法则。在某些病毒中的RNA自我复制(如烟草花叶病毒等)和在某些病毒中能以RNA为模板逆转录成DNA的过程(某些致癌病毒)是对中心法则的补充。
RNA的自我复制和逆转录过程:在病毒单独存在时是不能进行的, 只有寄生到寄主细胞中后才发生。逆转录酶是一种很重要的酶,他能以已知的mRNA为模板合成目的基因。在基因工程中是获得目的基因的重要手段
DNA的复制
DNA在DNA解旋酶的作用下两条链分离开,分别作为一个模板,在DNA聚合酶的作用下合成一条新链。
模板的3’端要有一段双链引物(primer),引物是引物酶(primase)协助合成的小段RNA。
![]()
DNA聚合酶从引物开始把适当的核甘酸5’->3’方向聚合上去,形成双链
从5‘端到3’端方向 5‘—>3’
![]()
DNA到mRNA转录

DNA到mRNA转录 主要功能剪去不表达的内含子
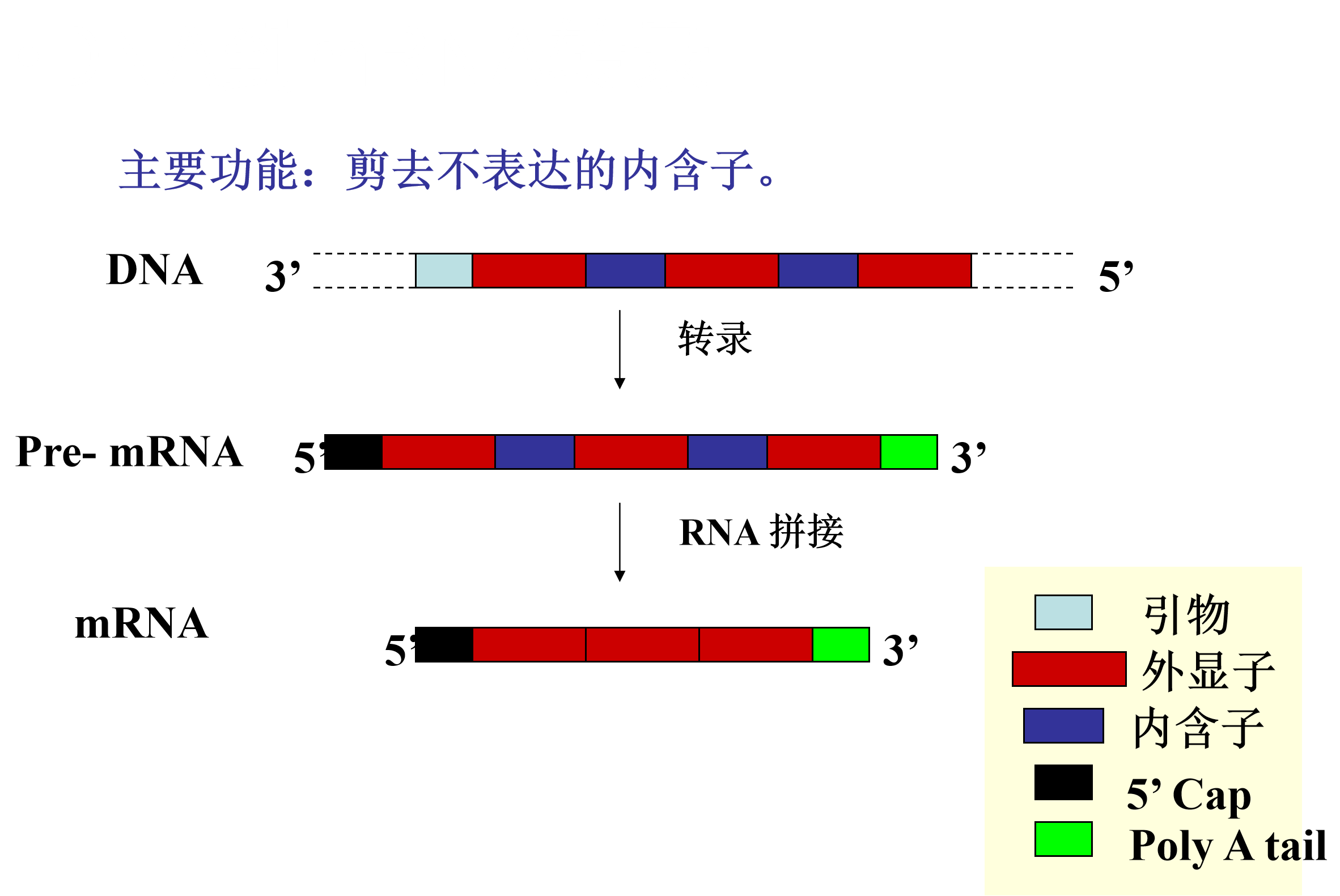
蛋白质的剪接
蛋白质的剪接:有些新生肽链要剪去中间一段,把两边连接起来,才变成成熟的功能蛋白质的过程。
被剪切掉的肽链称为内质(intein),留下的部分称为外质(extein)。
内质序列的N端大约有100个氨基酸,C端大约有50个氨基酸,构成剪接区。
这两个剪接区各自有一些保守的模体(motifs)
蛋白质的折叠
新生的肽链折叠为唯一的、特定的三维结构
折叠所需信息完全包含在氨基酸排列成的一维链中。(C.B.Anfinsen 1972)
折叠单元:由α螺旋和β片层组装成的紧凑折叠起来的单元;对蛋白质结构的分类和预测起重要作用;其种类极有限,可能不超过1000种。
结构域:蛋白质的氨基酸序列种有一些演化过程中最为保守的单元;一个结构域不能再划分为更小的结构域;一个蛋白质可含有一个或多个结构域。
突变与多态性

组 学
蛋白质组是细胞功能和状态的最直接描述,转录组成为研究基因表达
组学:通常指生物学中对各类研究对象(一般为生物分子)的集合所进行的系统性研究,而这些研究对象的集合被称为组学。例如,基因组学、蛋白质组学、转录组学、代谢物组学等。
转录组
转录组(transcriptome)广义上指某一生理条件下,细胞内所有转录产物的集合,包括信使RNA、核糖体RNA、转运RNA及非编码RNA;狭义上指所有mRNA的集合。
蛋白质是行使细胞功能的主要承担者,蛋白质组是细胞功能和状态的最直接描述,转录组成为研究基因表达的主要手段,转录组是连接基因组遗传信息与生物功能的蛋白质组的必然纽带,转录水平的调控是目前研究最多的,也是生物体最重要的调控方式。
蛋白质组
蛋白质组(Proteome)的概念最先由Marc Wilkins提出,指由一个基因组(Genome),或一个细胞、组织表达的所有蛋白质(protein). 蛋白质组的概念与基因组的概念有许多差别,它随着组织、甚至环境状态的不同而改变。 在转录时,一个基因可以多种mRNA形式剪接,一个蛋白质组不是一个基因组的直接产物,蛋白质组中蛋白质的数目有时可以超过基因组的数目。 蛋白质组学(Proteomics)处于早期“发育”状态,这个领域的专家否认它是单纯的方法学,就像基因组学一样,不是一个封闭的、概念化的稳定的知识体系,而是一个领域。
代谢组
代谢组(Metabolome)是指生物体内源性代谢物质的动态整体。而传统的代谢概念既包括生物合成,也包括生物分解,因此理论上代谢物应包括核酸、蛋白质、脂类生物大分子以及其他小分子代谢物质。但为了有别于基因组、转录组和蛋白质组,代谢组目前只涉及相对分子质量约小于1000的小分子代谢物质。
组学数据简介
表观遗传
表观遗传学(epigenetics)是研究基因的核苷酸序列
不发生改变的情况下,基因表达的可遗传的变化的一门遗传学分支学科。表观遗传的现象很多,已知的有DNA甲基化(DNA methylation),基因组印记(genomic imprinting),母体效应(maternal effects),基因沉默(gene silencing),核仁显性,休眠转座子激活和RNA编辑(RNA editing)等。
复杂生物网络
生物网络:在生物系统中用网络的形式表征基因、分子的调控以及相互作用关系,包含很多不同层面和不同组织形式的网络。最常见的有基因转录调控网络、生物代谢与信号转导网络和蛋白质相互作用网络等
影像组
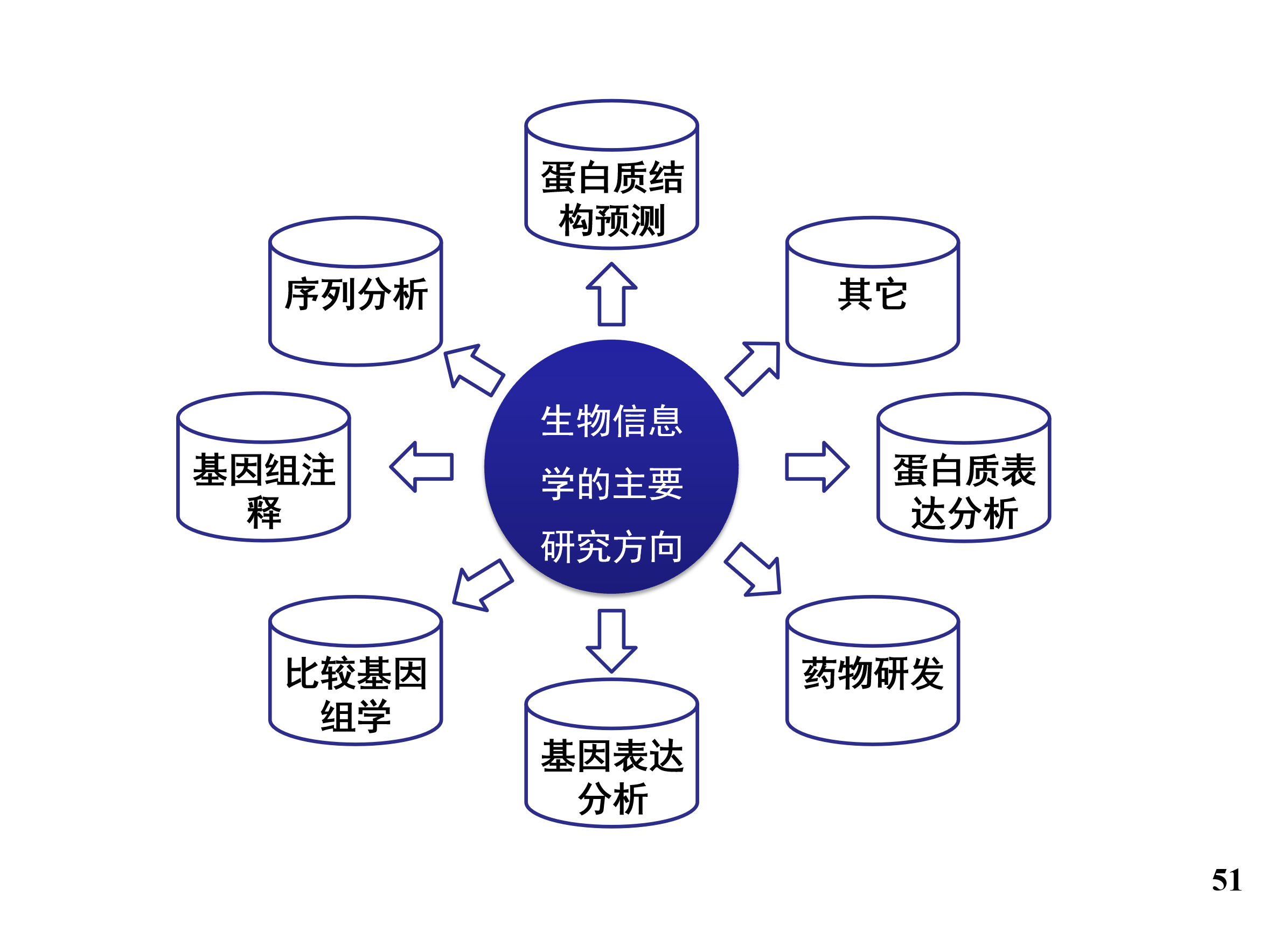
主要研究内容介绍
组学数据分析
基因组: 序列比对、改错、拼接、压缩……
转录组
代谢组
蛋白质组
微生物组学医学影像数据分析
医疗大数据分析
多序列比对
全局序列比对
动态规划算法(Dynamic Programming Algorithm)
分治法(Divide and Conquer Methods)
SP方法(Sum of Pairs Methods)
累进方法(Progressive Methods)
迭代方法(Iterative Methods)
遗传算法(Genetic Algorithms)
局部序列比对
由概形分析(Profile Analysis)
区块分析(Block Analysis)
统计学方法(Statistical Methods)
穿插内容:mooc内容山东大学
1.序列定义及分类:蛋白质序列&核酸序列
![]()
2. 序列相似性
数据库中的序列相似性搜索
对于一个蛋白质或核酸序列,你需要从序列数据库中找到与它相同或相似的序列。不可能再用眼睛去比较每一对序列,因为数据库中有太多序列。
UniProtKB下属的两个数据库

![]()
3. 一致度和相似度
用一致度和相似度这两个指标用来描述这两个序列有多相似
![]()
4. 替换记分矩阵
![]()
![]()
总结:【BLAST矩阵】 为DNA序列最广泛采用的矩阵
![]()
因此,BLOSUM后面跟一个小数字的矩阵适合用于比较相似度低的序列,也就是亲缘关系远的序列;
BLOSUM后面跟一个大数字的矩阵适合比较相似度高的序列,也就是亲缘关系近的序列。
PAM-250矩阵
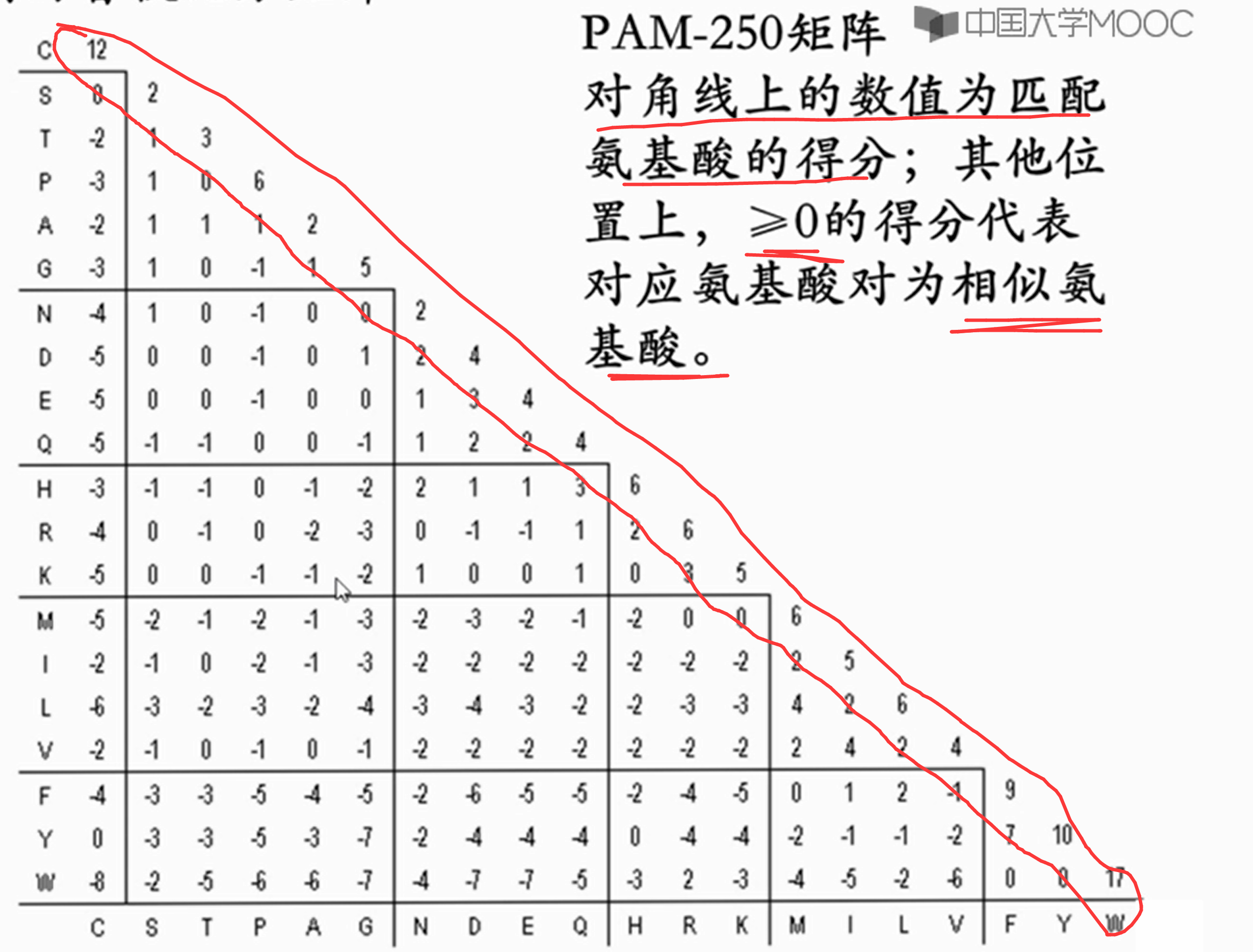
总结:【PAM矩阵】为蛋白质序列比较中最广泛使用的积分方法之一

现在我们总结一下到底是用PAM几,或者BLOSUME几。PAM1对应的氨基酸差异是1%,这是基础矩阵,由实际数据计算得出。而PAM11是由PAM1自乘11次得到的,他对应的氨基酸差异可不是11%,而是大约在**10%**左右。
同样,PAM80对应的差异也不是80%,而是在50%左右。如果你要比对的序列亲缘关系远,比如氨基酸差异在80%左右,那就得选PAM自乘次数非常多的矩阵,适合的是PAM246。但是现成的PAM矩阵也不是什么号的都有,只有几个关键号的。比如这个PAM246 就没有,有的是PAM250。
BLOSUME后面的号和PAM刚好相反,因为它对应的是序列的相似度。差异
在80%左右意味着相似度在20%左右,所以这个档次上的序列适合用的BLOSUM矩阵就是BLOSUM20。概括的说,PAM 后面的数体现的是序列的差异度,但不直接等于差异度,只是成对应关系而已;
BLOSUM后面的数体现是的序列的相似度并且直接等于相似度。所以我们看到,随着差异度的增大,适用的PAM 矩阵后面的编号是增大的,而BLOSUM 矩阵后面的编号是减小的。
![]()
其他2种蛋白质序列比对的替换记分矩阵
![]()
5.两个序列的长度相同计算一致度与相似度
seq1 : C L H K
seq2: C I H L
一致度:2/4=50%
相似度:需要知道 L 和 I 是否相似,K 和 L 是否相似
从BLOSUM-62矩阵中读一下
L: Leu I:Ile K:Lys
![]()
相似度:(2+1)/4=75%
6.两个序列的长度不同计算一致度与相似度
比较两个序列的方法:打点法&序列比对法
(1)打点法:
两个序列打点判断是否相似
![]()
在打点矩阵中,连续的对角线及对角线的平行线代表两条序列中相同的区域。这个矩阵中在主对角线位置上连续的红色的对角线说明这个位置对应的序列1的部分和序列2的部分是完全相同的,都是THEFA。此外,跟红对角线平行的蓝色平行线和绿色平行线,同样指出了序列1和序列2中两条相同的序列。也就是序列1和序列2中对应位置的TCAT,以及序列1和序列2中对应位置的AT
最后,我们放眼全局,红色的线和蓝色的线加起来基本上构成了一条主对角线。由此我们可以得出结论:序列1和序列2是比较相似的两条序列。
序列自己和自己打点寻找串联重复序列
![]()
![]()
用这种方法我们还可以快捷的发现序列中的串联重复序列以及重复的次数。我们只要数数在半个矩阵中包括主对角线在内的所有等距的平行线的个数,就可以知道重复的次数,而且最短的平行线对应的序列就是重复单元。短的串联复序列具有高度多态性,也就是说不同的个体间重复次数存在差异,而且这种差异在基因遗传过程中一般遵循孟德尔共显性遗传规律,所以快速查找某些特定的短的串联复序列的重复次数可以用于法医学的个体识别或亲子鉴定等领域。
打点法在线软件
最常用的Dotlet:https://myhits.sib.swiss/cgi-bin/dotlet

(2)序列比对法:
![]()
两条序列的比对就是把 s 和 t 这两个字符串上下排列起来,在某些位置插入空格,这些空
格叫空位(gap)。然后依次比较它们在每一个位置上字符的匹配情况,匹配的好,这个位置
就会得高分,匹配的不好,看看能不能左右错一错,或填上个空位,让附近的位置更好的匹
配在一起,从而使所有位置的得分之和尽可能的高。说白了,就是通过插入空位,让上下两
行中尽可能多的一致的和相似的字符对在一起。这不是随便摆摆看看就能完成的,需要使用
专门的序列比对算法。
全局比对算法:Needleman-Wunsch算法
全局比对输入值:
- 序列p和序列q,
- 替换记分矩阵,
- 空位罚分
![]()
如果不记得s(i,j)怎么计算了可以看看下面的链接
https://www.icourse163.org/learn/SDU-1001907001?tid=1461410447#/learn/content?type=detail&id=1237778652&cid=1257877487
s(1,1)的算法为例
w(i,j)对应当前位置的替换矩阵的得分
![]()
填满之后,右下角的分数就是整个全局比对最终的得分。
从这个位置**(最终得分)开始追溯箭头一直到左上角的零**
图中得分矩阵中标出的红色箭头是写出全局比对的唯一依据,追溯箭头是从右下角到左上角↖
但是写全局比对是从左上角开始。
![]()
全局比对(global alignment) :用于比较两个长度近似的序列
局部比对((local alignment):用于比较一长一短两条序列
局部比对算法:Smith-Waterman算法
![]()
与全局比对的区别:
1.得分矩阵初始值(0,0)仍然是0,但是第一行和第一列全是0
2.在选最大值时通过增加了第四个元素“0”,来达到比对局部效果,从s(1,1)开始要选择四个值中的最大值
3.如果0既不是从上面格,也不是从左边格,以及斜上格三个方向来的,而是来自于公式里面增加的“0”,则不用画箭头。
4.局部比对的得分不是在右下角,而是在整个得分矩阵中找最大值,这个最大值才是局部比对的最终得分。
5.追溯箭头不是从右下角到左下角,而是从刚刚找到的最大值开始追溯到没有箭头为止。
追溯箭头终止的位置也可以是得分矩阵中的任何一个位置
总结如何计算相似度
![]()
![]()
注意:不管两条序列长度是否相同,都要先对它们做全局比对。
都是要除以全局比对的长度来得到一致度和相似度
【学习笔记】生物信息学基础知识+序列比对初步了解(一)相关推荐
- php基础教学笔记,php学习笔记:基础知识
php学习笔记:基础知识 2.每行结尾不允许有多余的空格 3.确保文件的命名和调用大小写一致,是由于类Unix系统上面,对大小写是敏感的 4.方法名只允许由字母组成,下划线是不允许的,首字母要小写,其 ...
- 【学习笔记--FMCW基础知识】
学习笔记--FMCW基础知识 前言 mmWave测距原理 mmWave区分多个物体 mmWave的距离分辨率(Range Solution) mmWave的最大测量距离 前言 由于工作原因需要了解TI ...
- HTML5学习笔记 —— JavaScript基础知识
HTML5学习笔记 -- JavaScript基础知识 标签: html5javascriptweb前端 2017-05-11 21:51 883人阅读 评论(0) 收藏 举报 分类: JavaScr ...
- SAS学习笔记1——基础知识(库、PDV、变量选择、观测值排序、创建新变量
SAS学习笔记1--基础知识 1.逻辑库.临时库.永久库 2.数据步 2.1数据步语法 2.2 数据步的编译和执行过程 2.3变量的选择 2.3.1 keep和drop语句 2.4变量的重命名rena ...
- JS学习笔记——入门基础知识总结
JS入门基础知识总结1 前言 基础背景知识 一.产生历史: 二.特点: 三.应用方向: 四.Javascript组成: JavaScript书写使用方式 一.行内式(了解即可,项目中不使用,日常练习尽 ...
- 《UNIX 环境高级编程》学习笔记——UNIX 基础知识
UNIX环境高级编程--UNIX 基础知识 引言 UNIX 体系结构 登录 文件和目录 输入和输出 程序和进程 出错处理 用户标识 信号 时间值 系统调用和库函数 引言 所有操作系统都为它们所允许的程 ...
- 单片机学习笔记——微机基础知识
微机基础知识 微处理器,微机和单片机概念 微处理器的组成 一.运算器 主要寄存器 主要寄存器(IR),指令译码器(ID) 程序计数器(PC) 地址寄存器(AR) 二.控制器 存储器和输入输出接口 一. ...
- CAN总线学习笔记 | CAN基础知识介绍
一.CAN协议 1.1 CAN协议简介 CAN 是控制器局域网络 (Controller Area Network) 的简称,它是由研发和生产汽车电子产品著称的德国 BOSCH 公司开发的,并最终成为 ...
- WebGL学习笔记(基础知识篇)
WEBGL基础知识介绍 1.场景(scene) 场景如其名,即显示3D空间内物体的容器,就好比一个箱子是一个3D场景. 2.坐标系: webgl使用笛卡尔坐标系(宽度.高度和深度),我们也可以指定使用 ...
最新文章
- 011_logback中的SMTPAppender
- jQuery版本的网页开关灯、jQuery版本网页开关灯的另一种写法
- IT届常用单词读法纠正
- C++引用,常量优化,四种类型转换符
- java多线程和长连接,三方转换通信的实践(2)——数据库端服务程序
- cad如何导出jpg文件?
- 麒麟软件、麒麟操作系统、银河麒麟、中标麒麟、优麒麟、国产操作系统下载。
- flask中的csrf防御机制
- 关于AARRR用户增长漏斗模型,你究竟了解多少?
- linux关闭firefox进程,Firefox 68+ 怎样关闭多进程
- 万字拆解欧莱雅:百年美妆帝国的数字化远征
- 亮点前瞻 | 首届 ServerlesssDays · China 大会议程发布
- 逆波兰式-C++实现方法
- chrome谷歌翻译失效解决
- CleanMyMac2023Mac下载排行最好的清理工具
- 【MongoDB】——数据存储结构与基本数据类型
- 利用jquery实现双向数据绑定,网友:哎呦不错哦!
- 功能安全分级及参考资料
- 硬盘数据恢复 U盘数据恢复 移动硬盘数据恢复
- Web应用安全十大主动安全措施
热门文章
- 话里话外:抓大放小,从产销主流程入手的策略
- Linux-ssh: connect to host 192.168.1.161 port 22: Connection refused
- 线性代数系列(八)--基变换、左逆、右逆和伪逆
- dubbo SPI是不是在炫技?
- android 纯代码选项卡,纯javascript实现tab选项卡效果代码
- 互联网晚报 | 淄博政府回应经济酒店价格上千;多家银行向员工追薪;特斯拉上海储能超级工厂开始招聘...
- 随鼓点跃动的身心:从随身音频系统谈起
- 数据库中的创建、修改、删除
- lichee linux nfs,lichee开发板搭建nfs
- 数据挖掘竞赛-一起挖掘幸福感EDA